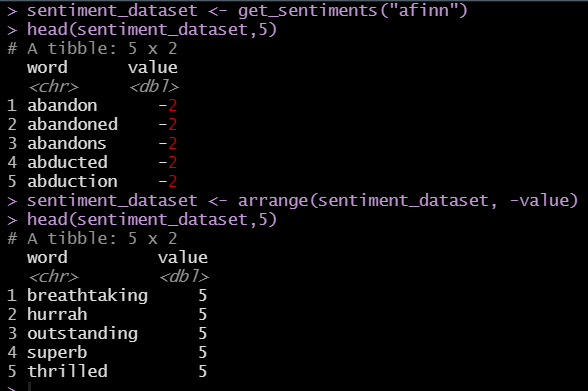
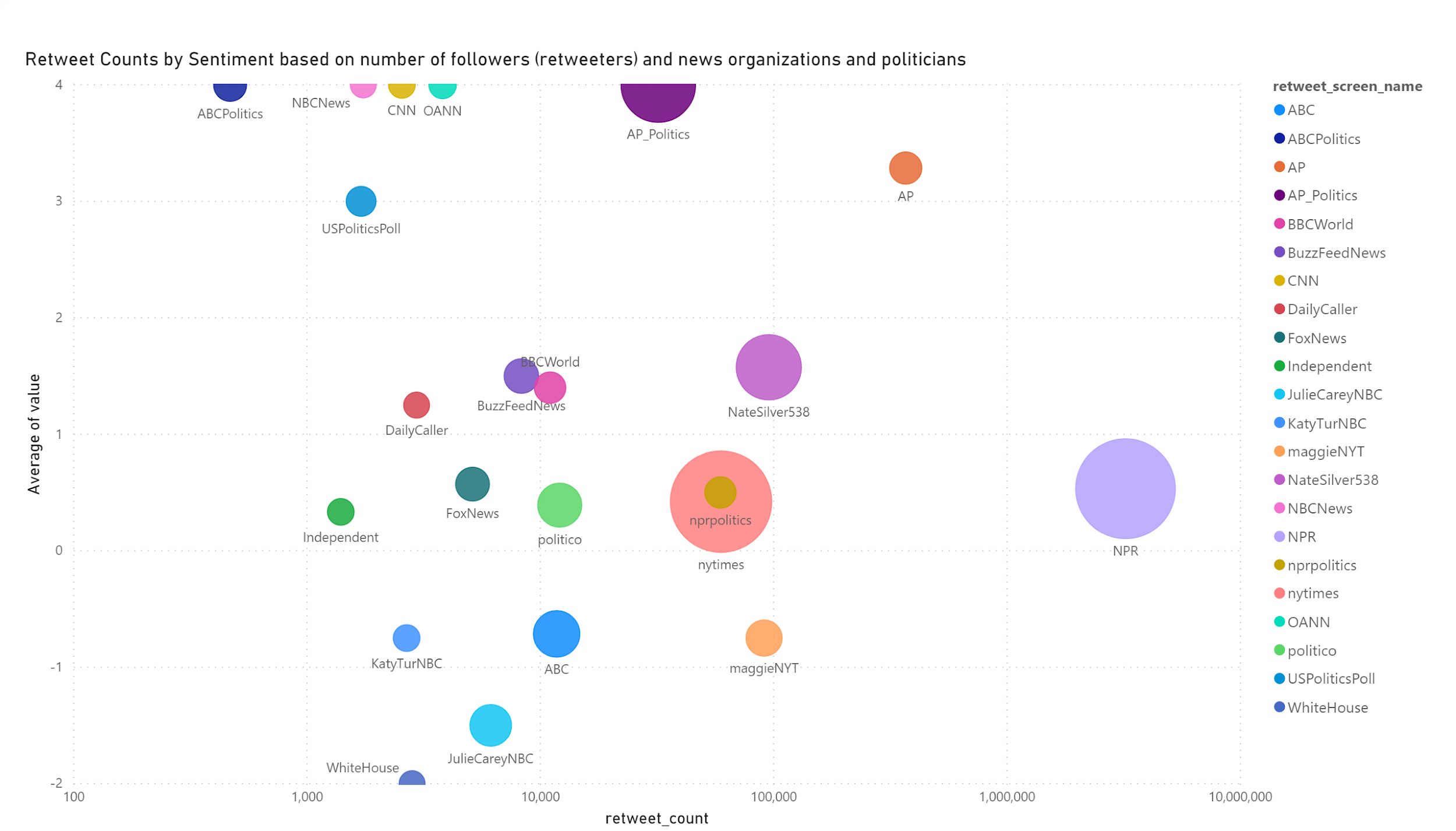
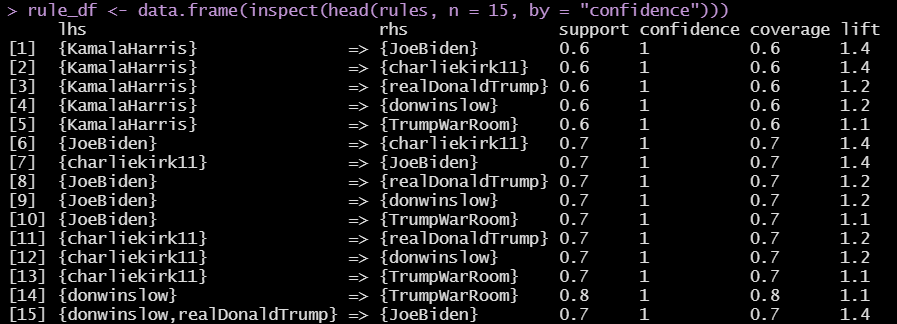
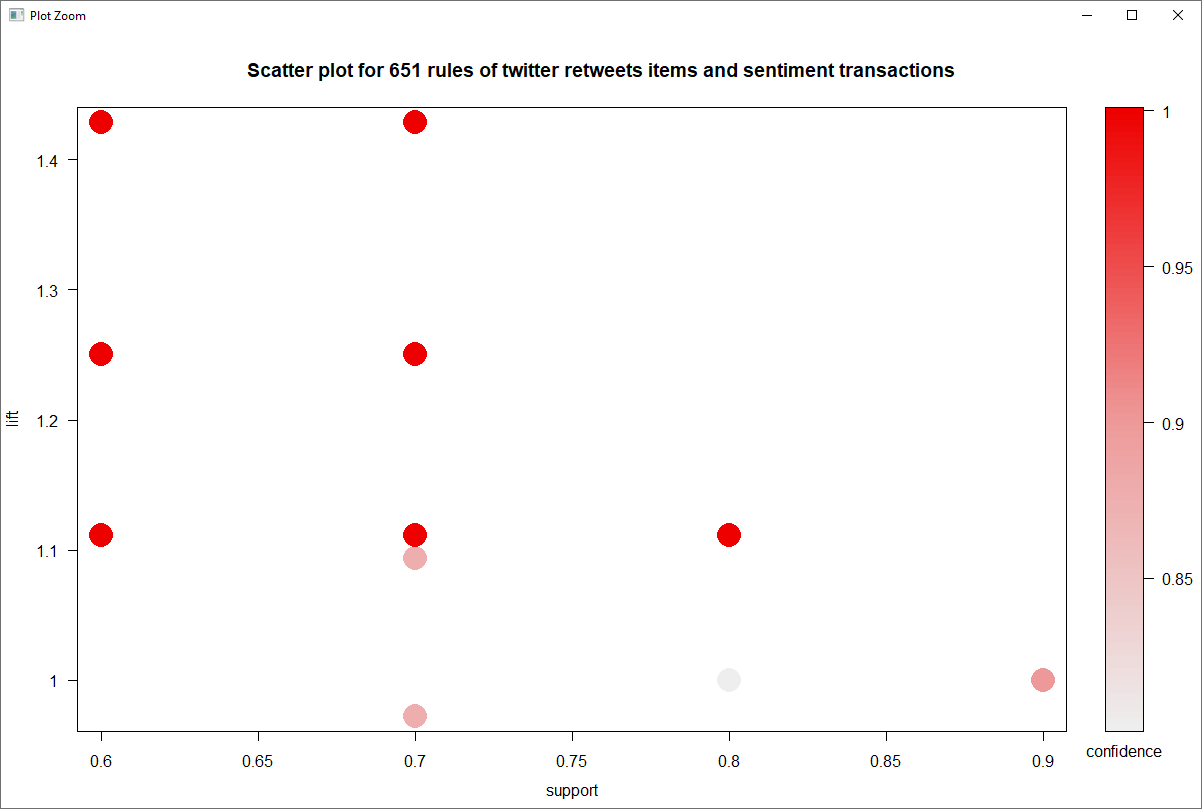
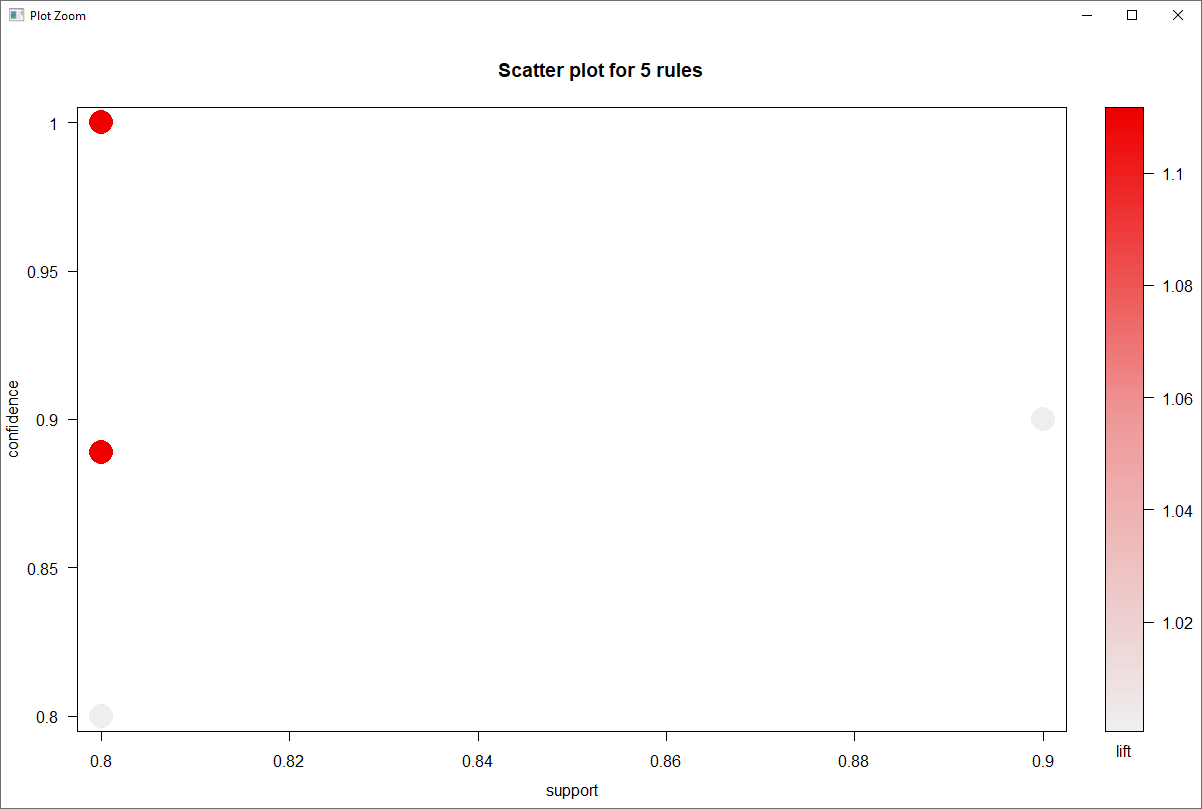
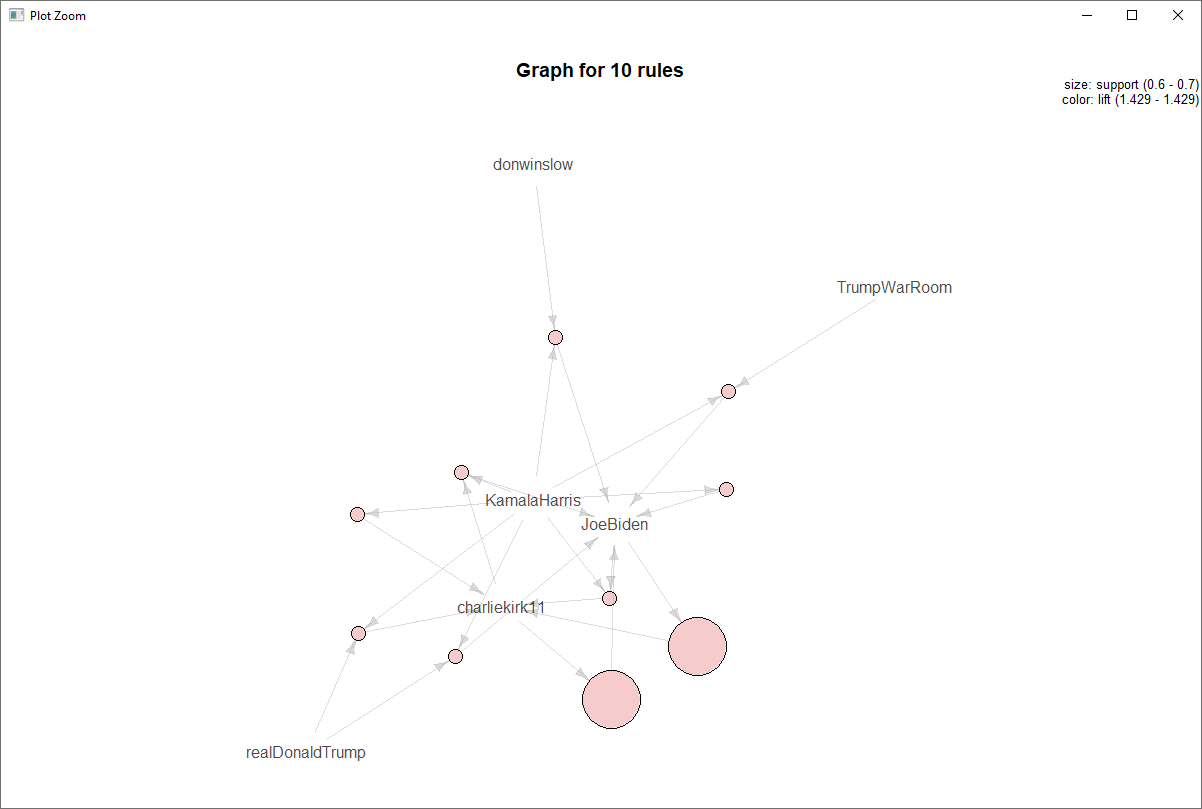
The U.S. Census Bureau American Community Survey application program interface (API) is a valuable programming interface for quickly and efficiently adding census data to your R code for analysis. It’s an extremely efficient way of doing data analysis in R without having to pull, format and organize census tables into a CSV or spreadsheet.
In order to use the census API, you will need to apply for a census API key, which takes only a few minutes. You then will need to install the key using the function census_api_key.
census_api_key('<key>', install=TRUE)Once the key is install, it doesn’t require to be installed again, unless it expires or you apply for new key. Going to the U.S. Census website and searching for the API is how to apply.
The get_acs R function gives you the ability to pull in multiple years and categories of survey information for demographic information including race, household income, marital status, employment, etc. It’s a power feature for analyzing survey and census data in tidy format.
ACS_2010 <- get_acs("state", year=2010, variables="S1702_C02_001", output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_2011 <- get_acs("state", variables="S1702_C02_001", year=2011, output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_2012 <- get_acs("state", variables="S1702_C02_001", year=2012, output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_2013 <- get_acs("state", variables="S1702_C02_001", year=2013, output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_2014 <- get_acs("state", variables="S1702_C02_001", year=2014, output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_2015 <- get_acs("state", variables="S1702_C02_001", year=2015, output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_2016 <- get_acs("state", variables="S1702_C02_001", year=2016, output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_2017 <- get_acs("state", variables="S1702_C02_001", year=2017, output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_S1702_C01_001_Family <- get_acs("county", variables="S1702_C01_001", output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_S1702_C01_013_Household_Work <- get_acs("county", variables="S1702_C01_013", output="tidy", geometry=TRUE) %>%
select(-moe)
ACS_household_work <- get_acs("county", variables="S1702_C01_013", output="tidy", geometry=TRUE) %>%
select(-moe)ACE variable data can be enumerated in a list with with descriptive names for easier classification. All variables are indicated by a document id (for example S1702_c01_018, S1702_C01_019, etc.).
ACS_Education <- get_acs("county", variables= c(nohighschool = "S1702_C01_018",
highschool = "S1702_C01_019",
somecollege = "S1702_C01_020",
collegeplus = "S1702_C01_021"),
output="tidy", geometry=TRUE) %>%
select(-moe)You can also choose to include margin of error (moe) or not. Visualizations can be in typical R GGplot library mode or included into maps. All ACS information includes IDs that categorizes survey data.
ACS_geo_2010 <- ACS_2010 %>%
select('GEOID','NAME','variable','estimate','geometry') %>%
filter(variable=='S1702_C02_001') %>%
group_by(GEOID, NAME) %>%
summarize(estimate = sum(estimate))
ACS_geo_2011 <- ACS_2011 %>%
select('GEOID','NAME','variable','estimate','geometry') %>%
filter(variable=='S1702_C02_001') %>%
group_by(GEOID, NAME) %>%
summarize(estimate = sum(estimate))
ACS_geo_2012 <- ACS_2012 %>%
select('GEOID','NAME','variable','estimate','geometry') %>%
filter(variable=='S1702_C02_001') %>%
group_by(GEOID, NAME) %>%
summarize(estimate = sum(estimate))
ACS_geo_2013 <- ACS_2013 %>%
select('GEOID','NAME','variable','estimate','geometry') %>%
filter(variable=='S1702_C02_001') %>%
group_by(GEOID, NAME) %>%
summarize(estimate = sum(estimate))
ACS_geo_2014 <- ACS_2014 %>%
select('GEOID','NAME','variable','estimate','geometry') %>%
filter(variable=='S1702_C02_001') %>%
group_by(GEOID, NAME) %>%
summarize(estimate = sum(estimate))
ACS_geo_2015 <- ACS_2015 %>%
select('GEOID','NAME','variable','estimate','geometry') %>%
filter(variable=='S1702_C02_001') %>%
group_by(GEOID, NAME) %>%
summarize(estimate = sum(estimate))
ACS_geo_2016 <- ACS_2016 %>%
select('GEOID','NAME','variable','estimate','geometry') %>%
filter(variable=='S1702_C02_001') %>%
group_by(GEOID, NAME) %>%
summarize(estimate = sum(estimate))
ACS_geo_2017 <- ACS_2017 %>%
select('GEOID','NAME','variable','estimate','geometry') %>%
filter(variable=='S1702_C02_001') %>%
group_by(GEOID, NAME) %>%
summarize(estimate = sum(estimate))Geographic Mapping of ACS Data
png(file="images/ACS_geo_2010.png")
tm_shape(ACS_geo_2010) + tm_polygons("estimate") + tm_layout(title.position=c("left","top"), title="% Poverty in U.S. Year: 2010 Post-Recession", asp=1)
dev.off()
png(file="images/ACS_geo_2011.png")
tm_shape(ACS_geo_2011) + tm_polygons("estimate") + tm_layout(title.position=c("left","top"), title="% Poverty in U.S. Year: 2011 Post-Recession", asp=1)
dev.off()
png(file="images/ACS_geo_2012.png")
tm_shape(ACS_geo_2012) + tm_polygons("estimate") + tm_layout(title.position=c("left","top"), title="% Poverty in U.S. Year: 2012 Post-Recession", asp=1)
dev.off()
png(file="images/ACS_geo_2013.png")
tm_shape(ACS_geo_2013) + tm_polygons("estimate") + tm_layout(title.position=c("left","top"), title="% Poverty in U.S. Year: 2013 Post-Recession", asp=1)
dev.off()
png(file="images/ACS_geo_2014.png")
tm_shape(ACS_geo_2014) + tm_polygons("estimate") + tm_layout(title.position=c("left","top"), title="% Poverty in U.S. Year: 2014 Post-Recession", asp=1)
dev.off()
png(file="images/ACS_geo_2015.png")
tm_shape(ACS_geo_2015) + tm_polygons("estimate") + tm_layout(title.position=c("left","top"), title="% Poverty in U.S. Year: 2015 Post-Recession", asp=1)
dev.off()
png(file="images/ACS_geo_2016.png")
tm_shape(ACS_geo_2016) + tm_polygons("estimate") + tm_layout(title.position=c("left","top"), title="% Poverty in U.S. Year: 2016 Post-Recession", asp=1)
dev.off()
png(file="images/ACS_geo_2017.png")
tm_shape(ACS_geo_2017) + tm_polygons("estimate") + tm_layout(title.position=c("left","top"), title="% Poverty in U.S. Year: 2017 Post-Recession", asp=1)
dev.off()